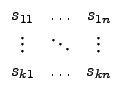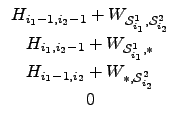Next: Error types and rates Up: Fundamentals of sequencing and Previous: Fundamentals of sequencing and Contents Index
A genome physically and chemically consists of tightly coiled strands of deoxyribonucleic acids (DNA) which is normally organised in a twisted double helix. The actual information contained in DNA is encoded by four different nitrogenous bases: Adenine (A), Cytosine (C), Guanine (G) and Thymine (T).
In a DNA helix, each base will always pair with its complementary base.
The bases are arranged along a sugar-phosphate backbone. The information needed to create any organism is specified by the sequence of bases - and the molecular structure defined by it - within the DNA. Refer to Bruce et al. (1994) for more information on working mechanisms of nucleic acids in general.
As will be explained later on, extracting DNA sequence information from cells is a tedious process involving many different steps. Each of these steps is prone to errors. Due to these errors, uncertainties remain once a sequence has been determined.
![]() b contains only those characters that are needed to represent the
discrete DNA sequence as it is found in cells.
b contains only those characters that are needed to represent the
discrete DNA sequence as it is found in cells.
![]() b = {A, C, G, T}
b = {A, C, G, T}
![]() n extends
n extends
![]() b with the character N (aNy) which shall stand for
bases that could not be determined more specifically in the analysis process.
b with the character N (aNy) which shall stand for
bases that could not be determined more specifically in the analysis process.
![]() n= {A, C, G, T, N}
n= {A, C, G, T, N}
![]() g extends
g extends
![]() n with the gap character ``*'' which implies
missing bases within a sequence
n with the gap character ``*'' which implies
missing bases within a sequence
![]() . ``*'' is also sometimes denoted as
padding character.
. ``*'' is also sometimes denoted as
padding character.
![]() g= {A, C, G, T, N, *}
g= {A, C, G, T, N, *}
With the help of the definition for the length of a sequence, the raw definition of a sequence can be refined.
![]()
![]()
The length of DNA sequence of organisms present on earth ranges from a few hundred kilobases up to several gigabases. For example, the bacterium Helicobacter pylori 26695 (GenBank accession number AE000511) has 1,667,867 bases (1.7 megabases) in its form as completed on September 7, 2001. In contrast, the human genome consists of approximately 3 gigabases.
But the gel or capillary electrophoresis technologies used to analyse DNA sequences can determine only about a maximum of 1,000 to 1,500 consequent bases, the high quality stretch with low error probabilities for the called bases often being around the first 400 to 500 bases. The problem to be solved is to find a method that allows the sequencing of long genomes, with the limiting factor that no subsequence to be analysed may be longer than 1 to 1.5kb.
![\includegraphics[width=\textwidth]{figures/shotgun}](img24.png) |
A simplified overview on the principles of the shotgun methods is given in figure 1. The DNA to be sequenced is first purified and amplified through cloning and then randomly fragmented into small parts, for example through sonication or the use of restriction enzymes. The fragments, also called inserts, are then sorted by size into so-called clone libraries of different sizes. For example, a genome sequencing project might use four clone libraries, where the fragments of the first library are approximately 2 kilobases in size, the second has 5 kilobases, the third 10 kilobases and the last 50 kilobases.
With the sensitivity levels of current sequencing technology, any type of nucleic acid sequence that is to be sequenced must first be amplified prior to its determination. This is done by inserting the fragment (insert) into a so-called cloning or sequencing vector and then inserting the resulting construct (a plasmid) into host cells - e.g. a bacterium like E. coli - for amplification as is shown in figure 2.
![\includegraphics[width=\textwidth]{figures/seqvector}](img25.png)
|
Once each vector/payload construct is amplified enough, a labelled polymerase replication process is started in the known sequencing vector part in forward and reverse direction as shown in figure 3. When using the chain termination dideoxy method published by Sanger et al. (1977), the replicates are terminated by dideoxynucleotides labelled with a fluorescent dye, a different dye for each terminating base. Each replicate will terminate at a different position of the fragment as the dideoxynucleotide prevents the polymerase from continuing the synthesis.
![\includegraphics[height=6cm]{figures/doublebarrel}](img26.png) |
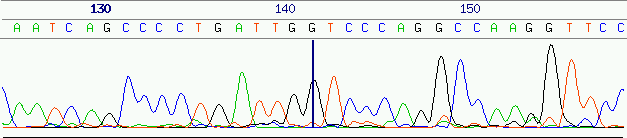
The combined replicates are then separated according to length in a gel or capillary electrophoresis process. The identity of a terminating base from a particular replicate can be detected by a two- or four-channel laser scanner as peak a in the fluorescence signal in the base specific channel. As short replicates will travel faster through the electrophoresis medium, the order of the replicates that pass the scanning mechanism also determines the order of the bases in each insert and thus the order of bases in the DNA fragment sequence.
Although trace signals are the most accurate representation of the sequences that have been analysed (see figure 4), they tend to be impractical as a basis for upcoming computational steps. Further on, genomes are a discrete ordered sequence of the four nucleotide bases. The trace signals must therefore be analysed to extract the original sequence.
Please refer to section 2.2 for a more in-depth discussion on the origin and the problems caused by wrong base calls.
The read sequences extracted from the forward polymerase replication (called forward reads) and the reverse polymerase replication (called reverse or complement reads) are all contained in the original DNA sequence of interest and are supposed to cover it completely in an overlapping way multiple times. But this is a working hypothesis only, based on stochastic assumptions. Although the fragments are assumed to be distributed uniformly along the DNA sequence (Idury and Waterman (1995)), chemical properties of biological sequences occasionally introduce a bias. In real-world projects, the reads will probably leave some gaps in the DNA sequence to be analysed, sometimes due to chemical properties of the sequence at this place (like coiling of DNA), sometimes just due to 'bad luck'. To reconstruct the original DNA sequence by computational means, the relative order and orientation 6 of the fragments must be determined. This inference is done using alignment algorithms.
| = | (1) |
It is clear that this definition of an alignment implies that all contained
sequences
![]() x must have the same lengths. Theoretically, one could use
the gap character (``*'') to pad the sequences to the left and/or to the
right and bring them to the desired lengths. But a ``*'' implies missing
characters and is often seen as an error of some sort. This is clearly not the
case for shotgun reads, as bases to the left or to the right of a sequence
have simply not been determined in the lab.
x must have the same lengths. Theoretically, one could use
the gap character (``*'') to pad the sequences to the left and/or to the
right and bring them to the desired lengths. But a ``*'' implies missing
characters and is often seen as an error of some sort. This is clearly not the
case for shotgun reads, as bases to the left or to the right of a sequence
have simply not been determined in the lab.
The alphabets
![]() x of the sequences
x of the sequences
![]() x are therefore extended by
the
x are therefore extended by
the ![]() character.
character.
| (3) | |||
| (4) | |||
| (5) |
Please note that
![]()
Normally, in alignments the ![]() character is often represented by ``.''
(point) or `` '' (blank). As this may lead sometimes to confusion for printed
text, the end-gap character will be represented by
character is often represented by ``.''
(point) or `` '' (blank). As this may lead sometimes to confusion for printed
text, the end-gap character will be represented by ![]() in plain text and
with a point or a blank in alignments. Please also note that
in plain text and
with a point or a blank in alignments. Please also note that
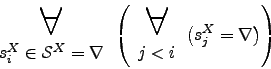
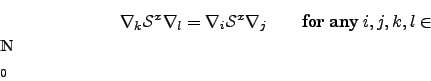
E.g.: ACGT = ACGT![]() =
= ![]()
![]()
![]() ACGT
ACGT![]()
Any sequence can therefore be brought to a desired length to fit into an alignment.
E.g.:
![]() X =
X = ![]()
![]()
![]() ACGT
ACGT![]()
![]() = 4
= 4
![]() = 8
= 8
As can be seen in equation 2.2 on page ![[*]](crossref.png) ,
each column in an alignment forms a k-tuple over the alphabet
,
each column in an alignment forms a k-tuple over the alphabet
![]() X. Two important numbers can be extracted from this k-tuple:
coverage and column score.
X. Two important numbers can be extracted from this k-tuple:
coverage and column score.
A scoring function is needed to asses the similarity of two or more aligned sequences, as the ultimate goal is to optimise the alignment.
| A | C | G | T | * | |
| A | 1 | -1 | -1 | -1 | -2 |
| C | -1 | 1 | -1 | -1 | -2 |
| G | -1 | -1 | 1 | -1 | -2 |
| T | -1 | -1 | -1 | 1 | -2 |
| * | -2 | -2 | -2 | -2 | 0 |
An example for a simple weight matrix W is given in table 1. Using this weight matrix, the score of an entire column can be calculated.
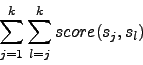
As an alignment consists of several columns, a method is needed to asses the quality of the total alignment using the column scores as reference.
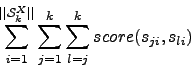
The shortest common superstring problem with strings containing no errors is NP-hard (Armen and Stein (1995)). It is a reasonable assumption that the same problem with strings (reads) containing errors cannot be simpler although this has not been proven formally.
E.g.: The two sequences TACGTCAATTAGATCTACT and CTACTGTA could be
globally aligned like this:
although this does not seem to make much sense as these two sequences are obviously not very similar when considering the entire length. For cases like this, local alignment methods are much more appropriate.
E.g.: The two sequences from the example above (TACGTCAATTAGATCTACT and CTACTGTA) can be locally aligned like this,
Now that alignments have been defined, the problem is still to calculate the alignments for a set of k given sequences. This problem has been solved in the 1970s and 80s by Needleman and Wunsch for global alignments by applying dynamic programming algorithms. Dynamic programming computes a sequence comparison and sequence alignment by comparing shorter subsequences first, so that their score can be made available in a table for the next longer subsequence comparison. Smith and Waterman extended this technique to local alignments.
Dynamic programming algorithms that locate optimal alignments of two sequences are central techniques for the comparison of biological sequences and have been extensively studied for the case of k = 2 sequences.
Equation 2.6 shows an example for a general 2-dimensional matrix
H, see also figure 26 on page
for the banding specialisation of the algorithm.
To calculate the value of Hi1, i2, it is only necessary to know the contents of the three predecessor cells (Hi1-1, i2-1, Hi1, i2-1, Hi1-1, i2).
While the multiple alignment problem is NP complete in the number of sequences, there is a well-known O(nk) dynamic programming algorithm for computing alignments among k sequences of average length n (Smith and Waterman (1981)). The problem can be solved by simply extending the dynamic programming recurrence for the basic problem: computing the values of matrix H in some topological ordering requires a total relative time of
While the extension to multiple sequences is conceptually quite straightforward, the obvious algorithm to compute the exact solution takes an amount of time exponential in k. It is therefore generally impractical in real world problems for k > 3 (Myers (1991)) sequences and - even when restricting the computation space by some means - certainly so for k > 4 (Gotoh (1993)).
Bastien Chevreux 2006-05-11