Next: Peculiarities of EST sequencing Up: Fundamentals of sequencing and Previous: Definitions and methods Contents Index
Depending on a multiple factors - ranging from clone preprocessing and
different dye-labelled terminators (or primers) to the type and length of gel
used during electrophoresis (see also Lario et al. (1997); Rosenblum et al. (1997)) - the
quality of the data gained along a single sequence substantially varies.
Current laboratory techniques can examine nucleotide sequence fragments
between 600 and 1300 bases long. In most cases there is a typical curve of
error rates to be observed (see Engle and Burks (1994); Lipshutz et al. (1994); Ewing et al. (1998); Engle and Burks (1993); Richterich (1998)): it starts
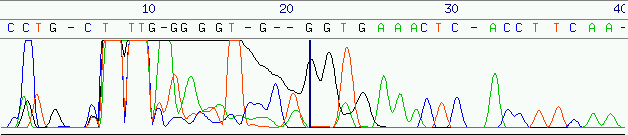
with a small stretch of low-quality bases (error rates between 3% and 8% for
the first 50 to 70 bases, see figure 5) followed by a stretch
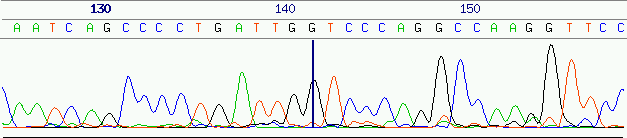
of high quality data (error rates ![]() 1% to 2% for the following 600 to 800
bases in good traces7,
figure 6), although it is nevertheless possible for
low-quality data to be present amidst a high quality stretch. As the
signal-to-noise ratio degrades towards the end of of a trace, the base quality
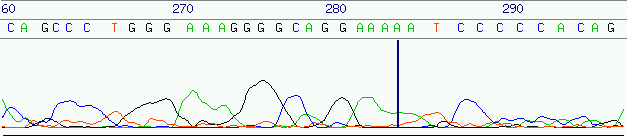
starts to deteriorate rapidly after a certain time with error rates ranging
from 2% up to over 10% and to 20% in the tail of the sequence like shown in
figure 7.
1% to 2% for the following 600 to 800
bases in good traces7,
figure 6), although it is nevertheless possible for
low-quality data to be present amidst a high quality stretch. As the
signal-to-noise ratio degrades towards the end of of a trace, the base quality
starts to deteriorate rapidly after a certain time with error rates ranging
from 2% up to over 10% and to 20% in the tail of the sequence like shown in
figure 7.
 |
 |
 |
Basically, there are three types of errors introduced into the data by electrophoresis and subsequent base-calling: insertions, deletions and mismatches. Insertions are wrongly called bases at places were there are none, deletions are bases that were not called in a sequence and mismatches represent wrongly called bases.8 These types of errors can be reduced by using improved chemistry (Lario et al. (1997); Rosenblum et al. (1997), by applying image processing algorithms (Sanders et al. (1991)) or by using different base calling algorithms (Berno (1996)).
Having a viable numerical estimate of the base quality has been a major advance achieved by Ewing et al. (1998) and Ewing and Green (1998) who presented an improved base caller that also gives probability values for the called bases expressed as confidence estimates.
The PHRED program was the first to transfer base error probabilities into a log-transformed value - also known as quality - to each called base.
Thus a quality of 40 would resort to the error probability of approximately 1 error in 10,000 bases.
One of the larger inconveniences is due to the method used to amplify small DNA clones which consist of adding an amplification vector and inserting the resulting construct into host cells (see also section 2.1). This vector/payload construct leads to an unpleasant consequence: any DNA sequence determined is likely to contain some part of the sequencing vector itself at the start - and sometimes the end - of the determined sequence. These stretches must of course be electronically removed as they do not belong to the target DNA that is to be sequenced. Unfortunately, the vector sequences are at the very front and rear of the sequence, which are the most error prone parts. Due to these errors, simple pattern matching algorithms often fail to recognise the sequencing vector completely.
The self-replication of the host-cells itself induces two further kind of errors: 1) errors in the base replication itself, which leads most of the time to small point mutations (SNPs, Single Nucleotide Polymorphisms) or 2) errors on a larger scale where the vector can ``loose'' its sequence payload, recombine with other plasmids or even recombine with some sequence parts of the host cell.
Please note that SNPs can observed both because of errors in the base-calling process and because of real sequence differences.
While infrequent errors on the SNP level do not pose a particularly difficult problem, a non-recognised recombination of the vector with any type of sequence from the replication host (a contamination) leads to completely wrong results in the downstream sequence analysis. Although the awareness to the problem of contamination has increased in the last years in the scientific community, a quick search in public databases for example still reveals an uncanny number of E. coli or known vector fragment stretches in sequence clones that were taken from human or rat chromosomes.
Another common type of biological problem due to random recombination of the vectors with other sequences is called chimera.
Bastien Chevreux 2006-05-11