Next: Results and discussion Up: Methods and Algorithms Previous: Iterative cycling Contents Index
For allowing the assembly of EST sequences and subsequent detection of SNP sites, the mira assembler was extended with some specialised algorithms: the miraEST modification of the standard assembler is specialised on using sequences gained by EST clone analysis to reliably detect and classify SNPs occurring in mRNA transcripts according to their SNP bases and strains (respectively cell types). Unlike other existing approaches like TRACE-DIFF (Bonfield et al. (1998)), polyphred (Nickerson et al. (2000)) or PTA (Paracel (2002c)), the method that was devised and implemented in the miraEST assembler does not first assemble all the sequences and then classify the SNPs. It rather uses information about potential SNP sites gained during the assembly to first cleanly assemble the transcript sequences by SNPs, strains, cell types and gene splice variants. Although it must be noted that not all SNPs or splice variants can be differentiated computationally: sequences with low transcript abundance and SNPs at sites having a low quality value cannot be differentiated. Also, splice variants that are a 100% subset of other variants will blend into their superset. Only in the last pass the resulting mRNA transcripts are assembled in a way that SNPs can be analysed, classified and reliably assigned to their corresponding mRNA transcriptome sequence.
The following sections describe both the problems and their solutions that were encountered respectively devised during the implementation and tests of the specialised EST assembly and SNP detection modules.
Knowing that some genes or gene families are expressed more often than others within a cell, it is clear that working with non-normalised EST libraries poses the risk that there might be a few hundred or even thousand very similar transcript sequences of a gene / gene family within the sequenced project like, e.g., cytochromes. On the other hand, rarely expressed genes might be represented just once or even not at all.
 |
The part of the mira assembler that showed to be the most susceptible to the increased coverage in EST projects was the pathfinder module with the ``width-first-depth-last n, m-recursive look-ahead strategy'' described in section 4.4.2. Understandably, a coverage of approximately 6 to 12 in genome projects with some repeat stretches scattered across the genome does by far not lead to graphs that are as dense as those build by, e.g., 1000 sequences that are very similar. The recursive nature of the path traversal algorithm led to disproportionate time and memory requirements. Different strategies were tried to tackle this problem. In the end, internal testing in the development phase showed that a time based cutoff-strategy combined with automatic search space reduction proved to be the most successful in terms of result quality, time and memory consumption. The time needed to traverse the connection graph is recorded for each recursion. If it surpasses a certain threshold, only the fraction of the possible targets corresponding to the ratio of time allowed versus time consumed is kept for the next recursion levels.
This strategy has, of course, a few drawbacks. First, its behaviour becomes that of a simple greedy algorithm for very large and dense graphs. Second, reproducibility is not guaranteed across different runs of the algorithm on different platforms or even on the same computer: the faster a computer is and the less other tasks are running, the more likely it will be that the search algorithm has enough time to analyse more solutions and not fall back to the quasi-greedy algorithm.
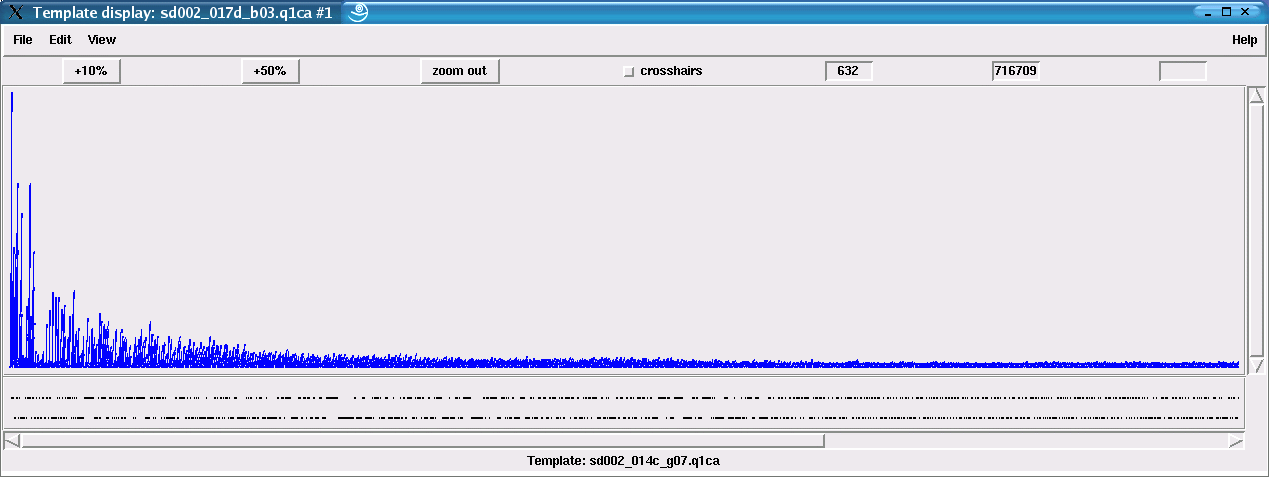
The best solution for human experts when doing this work manually is to search for patterns on a symbolic level in an mRNA transcript assembly to detect potential SNPs. Here again, the important factor is the observable circumstance that - normally - discrepancies in reads which cause a drop in the alignment quality do not accumulate at specific column positions. Sequences from different mRNAs, however, may show column discrepancies between bases of different reads that simply cannot be edited away: these are the potential SNP sites. See figure 41 for such a typical real life example.
![\includegraphics[width=\textwidth]{figures/sponge_c1_compact_all3t}](img169.png) |
Adjudicating now whether discrepancies between similar EST sequences are significant - and thus a polymorphism - or not relies therefore much more on the underlying traces and their quality than for genome assembly.
Based on this, the symbolic pattern recognition methods developed for recognition of column discrepancies in repeat alignments in section 4.6.3 were expanded to allow detection of SNPs. The algorithms developed base on the same working principles, i.e., the most important criterion are the group qualities that can be calculated for different bases in a column. Under those circumstances, even a discrepancy caused by a single base in a single column of an alignment can be seen as a hint for a SNP site, i.e., if the base probability values of the bases in the immediate area are high and the signal traces do not allow an alternative sequencing error hypothesis. The bases allowing discrimination of reads belonging to different mRNA transcripts will be marked as possible SNP marker bases (PRMB) in an intermediate first pass by the assembler. Technically speaking, the bases tagged as PRMB in this pass are - of course - not marker for possible repeats but marker for possible SNPs and should be named possible SNP marker base (PSMB). However, their function is exactly the same and handled by exactly the same routines.32
 |
 |
Basically, one can differentiate between three distinct types of single nucleotide polymorphisms (SNP) when analysing transcripts from one or several organisms, strains or cell types.
During an optional last assembly pass, the miraEST assembler will merge almost identical - strain and SNP separated - transcriptome sequences from the previous passes for a last alignment. Such an alignment shows SNP differences between the mRNA sequence transcripts. The transcript sequences used for this final assembly stage will be precisely classified and assembled at least by SNP types and - if the information was present - by organism / strain / cell type in the previous passes. Consequently, it is reasonable to assume that the transcript sequences used at this stage are pristine, that is, they code existing proteins.
It is important to note that this step, like the whole process performed by miraEST, is still an assembly and not a clustering step, i.e., sequences composed by different exon structures or which contain large indels will not be assembled. The results obtained here are nevertheless important in a sense that they allow analysis and classification of the SNP types of nearly identical mRNA sequences which occur in one or several sequencing assembly projects.
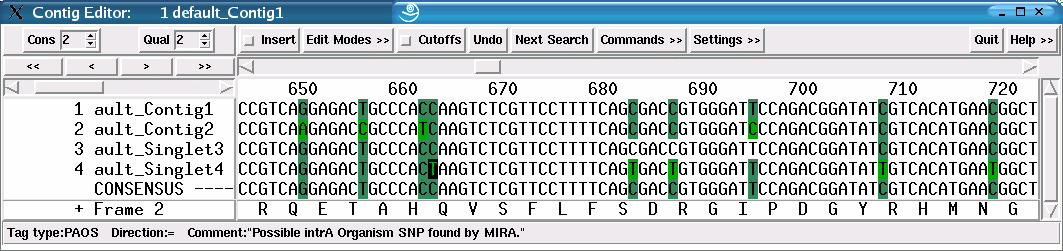
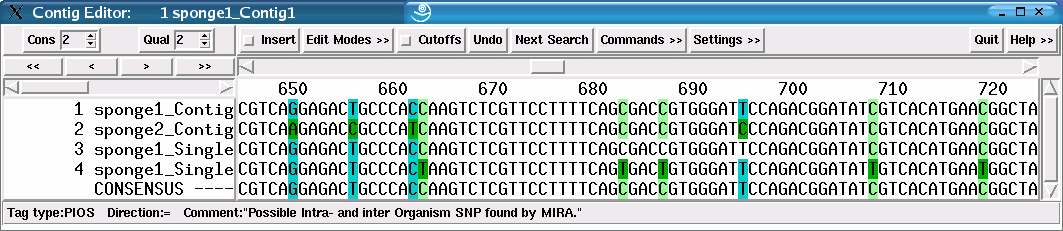
Figure 43 illustrates the assembly of two strains. SNPs are classified into all three categories, the example figure showing two of them (PAOS and PIOS). Sequences without strain information will also have the bases tagged, but only as PAOS as they will be assigned to a default strain. Comparing figures 42 and 43 exemplarily shows the differences in classification with and without supplied strain data. Figure 42 shows the assembly of one strain, each SNP is therefore classified as as PAOS. Figure 43 shows the simulated assembly of two strains. SNPs can now be classified into all three categories, the example figure shows two of them (PAOS and PIOS).
Poly-base stretches of As or Ts marking the end of translation are left as help for visual analysis, but are not used computationally to decide whether a position contains a SNP or not.