Next: Methods and Algorithms Up: Assembly strategies Previous: Existing strategies Contents Index
A multi-phased concept has been worked out to have the assembler perform the difficult task of optimal shotgun sequence alignments. Different authors have proposed different sets of acceptance criteria for the optimality of an alignment (an overview is given by Chan et al. (1992)). Traditionally (Myers (1995)), ''the objective of this [assembly] problem has been to produce the shortest string that contains all the fragments as substrings, but in case of repetitive target sequences this objective produces answers that are overcompressed.''
Overcompression can be compensated in part by using special cloning and sequencing techniques like taking different clone template insert sizes and using this information for a pre-assembly coverage analysis like it was done for the sequencing of the human genome by Celera company (Venter et al. (2001)). However, not all genomic or EST sequencing projects use this technique. Furthermore, although EST clone library projects can use dual end clone sequencing techniques, this information is of little help as mRNAs rarely surpass a few thousand bases in length. To make the matter worse, certain genes - or even complete gene families - in non-normalised EST clone libraries can be disproportionally expressed and sequenced more often than others (e.g. cytochromes). This results in more mRNA clones of these genes and thus more sequence fragments of these sequences.
Searching alignments by suffix trees and analysing the trees to deduce possible repeat resolving strategies was discussed several times in the past. But, as Ma et al. (2002) noted, suffix tree approaches suffer from two main problems: they are not efficient in handling mismatches and suffer from large space requirements. The later problem is gradually being addressed by more efficient algorithms like MUMmer2 and NUCmer (Delcher et al. (2002)) and - trivial but still important to note - technology advances that nowadays allow relatively inexpensive computers to have several gigabytes of memory.
Another method to handle repeats consists of reducing the fragment assembly problem to a classical variation of the Eulerian superpath problem (see Pevzner and Tang (2001)) which was extended in Pevzner et al. (2001) to use template insert size information14generated by clone-end sequencing. Still other methods like the one by Otu and Sayood (2003) used a very different strategy with divide-and-conquer approach by computing average mutual overlap information in their fragment assembly algorithm to simultaneously solve the overlap, layout and consensus phases of an assembly.
Lee et al. (2002) introduced heuristics using partial order graph to use pairwise dynamic programming for multiple sequence alignments to resolve problems posed by repetitive sequences and overcompression. One shortcoming of this approach was that it turned out to be not immune to order-dependency effects in the constructed alignments.
In the end, all this means that the overcompression criterion is only partly useful for tracking misassemblies in genomic sequencing and not useful at all for detecting misassemblies in EST sequencing projects. As a result to this, the strategy conceived was the one of the least number of unexplainable errors present in an assembly to be optimal.
 |
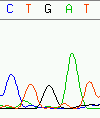 |
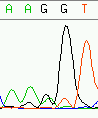
Unexplainable error are errors in an assembly where evidence in the trace data does not permit to correct wrongly called bases of the offending read toward a consensus. Figure 12 shows a simple case of an explainable error - which almost certainly will be edited away - while figure 13 pictures an unexplainable error in an assembly. Discrimination possibilities between base calling errors and real discrepancies is the main difference to using simple quality values as discerning criterion only: quality values do not offer alternatives to the called base. Regardless of the possible quality of a base, trace data always offers this possibility.
So, a novelty of the new assembly strategy is that the assembler has been combined with some capabilities of an automatic editor. Both the assembler and the automatic editor are separate programs and can be run separately, but the tasks of assembly and finishing can be viewed to be closely related enough for both parts to include routines from each other (see also Pfisterer and Wetter (1999); Chevreux et al. (1999)). In this process, the assembler gains the ability to perform signal analysis on partly assembled data which helps to reduce misassemblies especially in problematic regions like standard repeats - e.g. ALUs, LINEs, MERs, REPT etc., see Bruce et al. (1994) for more information on these - where simple base qualities alone could not help. Analysing trace data at precise positions with a given hypothesis 'in mind'15 is a substantial advantage of a signal analysis aided assembler compared with a 'sequential base caller and assembler' strategy, especially while discriminating alternative solutions during the assembly process. In return, the automatic finisher gains the ability to use alignment routines provided by the assembler.
Another important improvement of the developed assembly strategy that differentiates it from simpler alignment algorithms is the possibility to perform post-assembly validation checks on the resulting contigs. These checks are run to find misassembled reads and remove them from the assembly. Most often, these misassemblies are due to repetitive sequences that might differ in only one or two percent on stretches as long as several hundred bases. This is something almost impossible for alignment algorithms to discern correctly as these algorithms are specifically designed to work with slightly erroneous data. So, repetitive sequences still might get wrongly assembled together a first time. Fortunately, these repeats produce error patterns in an assembly that can be searched for and recognised by pattern recognition algorithms. These algorithms are helped by the fact that the automatic editor will have edited away most of the trivial base calling errors. The remaining error patterns can be compared to typical repeat error patterns and the offending reads can be tagged as repetitive and marked for a subsequent assembly iteration.
![\includegraphics[width=\textwidth]{figures/cyclemira2}](img78.png) |
Figure 14 depicts an approximate overview of the developed strategy as well as the different phases involved while assembling a project:
As can be seen, the overall structure of the assembly strategy chosen - i.e. the iterative nature of its design and the steps concentrating on high quality parts first - reflects the needs imposed to it by the aims set for this thesis: identifying repeats to avoid misassemblies and decrease the error rate of the final consensus sequence. Each subpart of the assembly strategy is embedded into the overall scheme, which in turn means that, sometimes, algorithms show unexpected - and mostly undesired - side effects when used within the whole system. The next chapter therefore goes into the details of those algorithms, explains the inner working mechanisms of each subpart and how they are connected to each other.
![\includegraphics[width=\textwidth]{figures/ourass}](img77.png)