Next: Conclusion and outlook Up: Results and discussion Previous: EST assembly Contents Index
Very early in the development process it was discovered that using high quality sequence data first in the assembly process was a very viable way to proceed as it substantially reduced computing time. This permitted to reinvest this saved time into other algorithms that increased the actual quality of the final results: resolving detected misassembly conflicts, analysis and detection of previously unknown relevant sequence features (e.g. repeat marker bases, SNPs, etc.) and detection and elimination of conflicts caused by misassemblies. The ever increasing computing power permitted the design of exact iterative algorithms instead on relying on makeshift algorithms when assembly problems occurred. That is, it was a clear choice not to trade off quality for speed when the loss in quality was deemed to be substantial. Furthermore, integrating an automated trace editor into the assembly process was the correct choice as results showed that spurious base-calling errors are reliably detected and removed in an alignment and the assembler can also use the trace analysis routines to perform in-depth and multi-level analysis on problematic regions in alignments.
Presently no other assembly system - be it for genomic or transcript data - contains a comparable mix of algorithms that enables the assembler to dependably detect by itself and use the information about special base positions that differentiate between repetitive stretches within sequences as is the case for repeat marker bases (RMB) in genomic assemblies or single nucleotide polymorphisms (SNPs) bases in EST assemblies. Reiterating the stance regarding the importance of discovering such base positions during the assembly: they allow the assembler to perform a reliable separation of almost identical sequences - which may ultimately differ only in one single position within two single sequences - into their true original genomic sequence or transcriptome. This is significantly more sensitive and specific than other methods like relying on base qualities alone (PHRAP) or the one presented by Tammi et al. (2002), which needs at least two differences in reads to distinguish them from sequencing errors.
Additionally, corrections performed by the integrated automatic editor resolve errors in alignments produced by base-calling problems. This makes RMB or SNP detection much less vulnerable to sequence specific electrophoresis glitches and base-calling errors as is the case for, e.g., the AG-problem known with the ABI 373 and 377 machines where a G preceeded by an A is often unincisive or only weakly pronounced.
The miraEST assembler was developed concurrently to the mira version for genome sequences presented in Chevreux et al. (2000,1999), which enabled to use basic algorithms for both branches of the assembly system. This also allowed to concentrate on developing and improving those algorithms that are specifically needed to tackle the slightly different assembly problems of genome and EST sequences once the basic facilities were in place.
In contrast to other assemblers or SNP detection programs - like phrap, gap4, pga/pta, the TGICL system, polyphred or autoSNP - the approach devised uses strict separation of sequences according to the real signals in the trace chromatograms. As the results presented in this chapter have shown, this is a reliable way to ensure that the consensus sequences produced as result correspond to the real genome or transcriptome sequence.
This method permits to use these results directly for the design of further investigative studies with high quality and precision requirements like, e.g. the design of oligo probes for specific SNP detection in clinical micro-array hybridisation screening experiments.
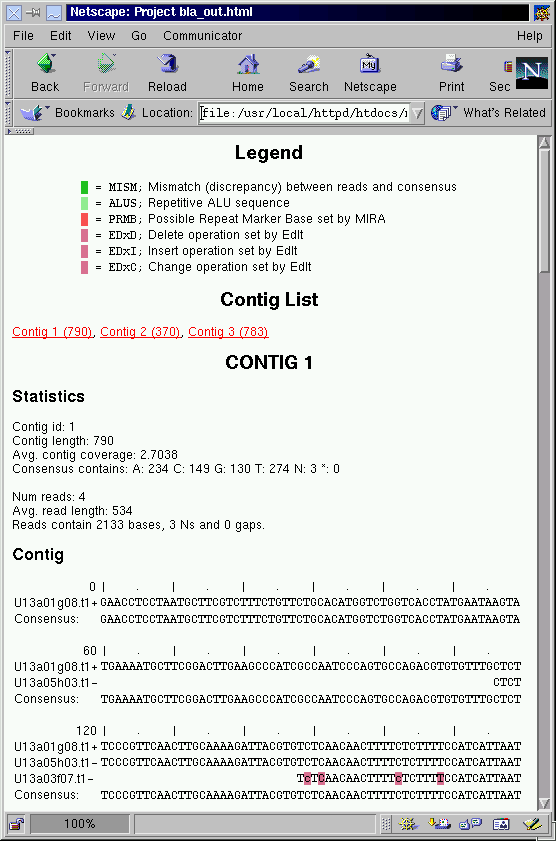
The possibility to export the assembled projects together with the analysis of RMB or SNP sites to a variety of standard formats, e.g. gap4 directed assembly, phrap .ace, or even simple HTML pages as shown in figure 44 opens the door to visual inspection of the results as well as integrating the tool into more complex and semi-automated to automated laboratory workflows.