| | Home | Projects | Images | Miscellaneous | Contact | Sitemap |
| Home :: Projects :: MIRA :: Examples :: Genome |
At the time of this writing (13.10.2003), the estimated 0.9 megabase bacterium Neorickettsia Sennetsu (Miyayama strain) was not completed and published by the TIGR center. Only the raw traces were submitted to the NCBI trace repository.
I downloaded the 12833 available traces along with the ancillary XML information file (which contained useful clipping information) and assembled this project with the V2.2.4 version of MIRA. I used standard setting, only telling the assembler to load the data from FASTA files, merge it with ancillary information present in the XML trace info file (clippings etc.) and use the SCF files as information for the integrated automatic editor.
The image below gives a short overview what the assembler did with the data and how it is presented in the Staden package.
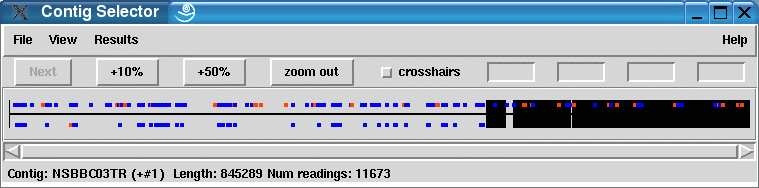 |
The screenshot shows the contig selector of the Staden package with the assembled NSM project loaded. What one can see is an almost complete assembly: the main contig (the long black line on the left side) has 845289 base pairs, the second smallest contig 15145 base pairs and the third largest contig has 4002 base pairs.
The rest of the contigs are smaller in size: 3kb and rapidly decreasing. The number of sequences in a contig also decreases rapidly until reaching single-read "contigs" (singlets)..
The dark blue and orange spots are tags set by the MIRA assembler representing weak repetitive marker bases (WRMBs) and probable repeat marker bases (PRMBs) respectively. These tags show the positions and the reads where the assembly algorithms identified potential high-quality mismatches leading to trouble. Some of the contradicting reads could be assembled at other positions, other contradictions could only be "resolved" by leaving out the most intrigueing reads.
An integrated automatic editor
The integrated automatic editor of the MIRA assembler helps to clarify potentially problematic situations for the pattern matcher of the assembly system. The image below shows a typical problem occuring during the assembly: a cluster of sequences differs in only very few bases from each other. The assembly engine identified theses and tagged them with an (orange) warning tag, while the integrated editor edited away mismatching bases according to the underlying trace signals.
The result is a clear match / mismatch pattern that identifies the bases that differentiate between several very similar repetitive regions in a genome
 |