Bastien Chevreux, March 2009.
Please comment in the SeqAnswers forum at http://seqanswers.com/forums/showthread.php?t=1353 or send mail directly to me (bach@chevreux.org).
Data from the Illumina GAI and GAII analyzer. 36mers (end of 2007) and 40mers (beginning of 2009) are affected, other lengths probably also. The data was generated by different sequencing labs / companies in Europe and the US, this therefore seems to be a lab-independent problem.
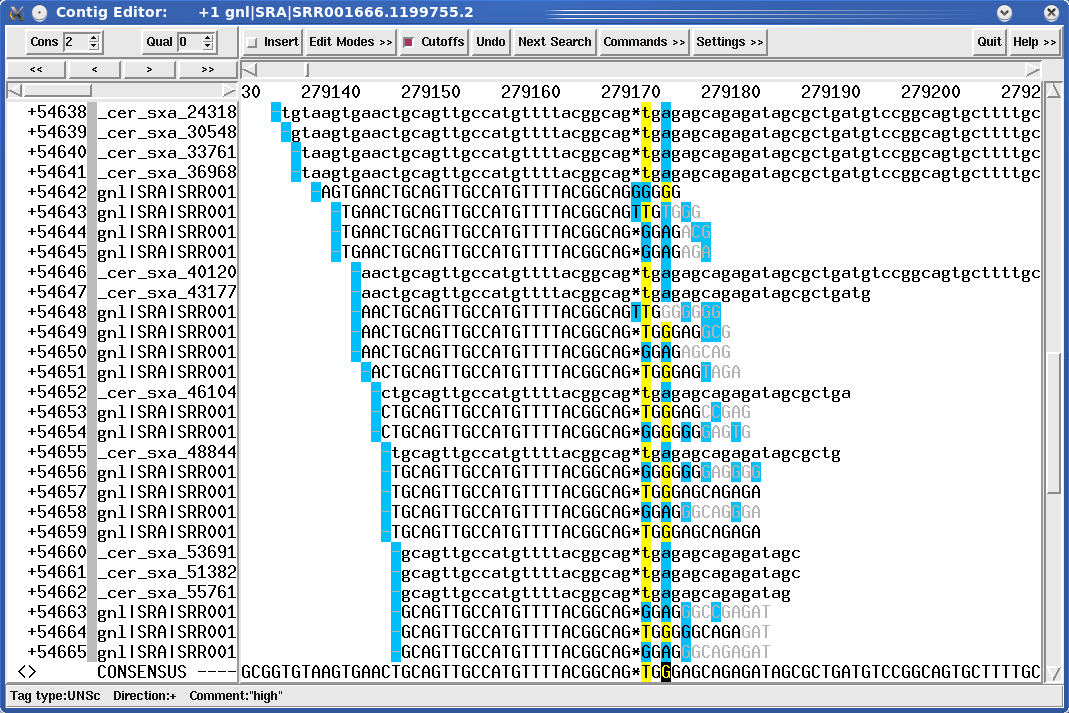
Certain errors in Solexa sequencing data apparently occur more often directly after GGC.G in 5' to 3' sequencing direction (the dot standing for any base). On a broader scope, GG..G is at risk. Quality values are usually very high in affected reads, no possibility for a quality clipping algorithm to catch this kind of error.
There are probably other factors as fortunately this does not occur everywhere in the genomes I'm working on, but when this problem strikes, it easily affects 1/3 to 1/2 of the reads, sometimes more. Also, the problem is more likely to occur in the second half of the read than in the first half.
I currently have no good explanation what could cause these errors.
Illustration 1: The true sequence is ggcagT and not ggcagG as a lot of reads show. Note that the sequence data in light grey is normally not shown as this was automatically detected to be bad sequence data and clipped away by MIRA.
This is something I found out recently but did not find described anywhere, so I've set up an example with public data (Ecoli K12 MG1655) so that others can have a look at and comment regarding possible causes. If you know or find out something, please feel free to discuss this on SeqAnswers (http://seqanswers.com/forums/showthread.php?t=1353).
Note that in this example I'll use only a subset of 7.4 million Solexa reads as MIRA is quite greedy with memory. You will still need a machine with 10 to 12 GiB RAM and some 2 to 6 GiB free swap (or more RAM :-)
In this walkthrough I'll use gap4 from the Staden package to look at the results. You should be able to use other viewers (consed or Mosaik or others), but if you want to follow this walkthrough line by line, you will need to have a working gap4 (http://sourceforge.net/project/showfiles.php?group_id=100316&package_id=107815) and also the caf2gap converter from the CAFTOOLS package at the Sanger Centre (ftp://ftp.sanger.ac.uk/pub/PRODUCTION_SOFTWARE/src/caftools-2.0.2.tar.gz).
Please download the 64 bit version of MIRA (currently 2.9.43) from http://chevreux.org/mira_downloads.html
Unpack the file and install the executables somewhere in your path. Or, later in this walkthrough, run it from where it was unpacked.
Change to a directory on a filesystem with at least 13 GiB free
mkdir mg1655
mkdir mg1655/origdata
mkdir mg1655/data
Download the following data from the NCBI
http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nuccore&qty=1&c_start=1&list_uids=NC_000913&uids=&dopt=gb&dispmax=5&sendto=&fmt_mask=504&less_feat=504&from=begin&to=end&extrafeatpresent=1&ef_MGC=16&ef_HPRD=32&ef_STS=64&ef_tRNA=128&ef_microRNA=256&ef_Exon=512
Save
it as “mg1655_backbone_in.gbf” into the mg1655/origdata
directory
http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?cmd=search&exp=SRX000430&run=SRR001666&m=search&s=seq
Select
“FASTA” format, click on “Save” and move the downloaded
data to mg1655/origdata (name it whatever you like, this
walkthrough uses mg1655_sxa_downloaded.fasta.gz
Note 1: for
this example I don't bother to download the quality data. With or
without quality data, the results are the same.
Note 2: this
data was deposited by Illumina
cd mg1655/origdata
gunzip mg1655_sxa_downloaded.fasta.gz
cd ../data
split -b 795m ../origdata/mg1655_sxa_downloaded.fasta.gz
mv xaa mg1655_in.solexa.fasta
rm xab
grep ">" mg1655_in.solexa.fasta | cut -f 1 -d ' ' | sed -e 's/>//' -e 's/$/ mg1655_sxa/' >mg1655_straindata_in.txt
ln -s ../origdata/mg1655_backbone_in.gbf .
cd ..
mkdir assemblies
mkdir assemblies/test1
cd assemblies/test1
lndir ../../data
mira
--project=mg1655 --job=genome,mapping,accurate,solexa
-SB:lb=yes:bft=gbf:bsn=mg1655_genbank:lsd=yes -AS:nop=1
SOLEXA_SETTINGS -AS:bdq=30 >&log_assembly.txt
Go
fetch a coffee or do something else in the next 45 minutes (if you
have a Core i7 940, might be slower or faster for your machine)
As I wrote: I'll use gap4. You can use other viewers which can load CAF or ACE data.
cd mg1655_d_results
caf2gap -project assembly -ace mg1655_out.caf >&/dev/null
gap4 ASSEMBLY.0
Open the contig editor and scroll to position 279170 which shows the example from the screen shot from the beginning of this text.
To find other places
where there's a GGC.G problem, use the search function from gap4 and
search for the following tags (which are set by MIRA): SROc, UNSc,
SRMc
Note that you need to make gap4 (or consed or eventually
other viewers) aware of the tags set by MIRA, Please read the README
in the support directory of the MIRA package to see how this is
done.
Note that the SROc normally shows true valid SNPs between strains, but most of the tags in this example are false positives due to the GGC.G problem ... and due to the fact that the GenBank sequence is a really, really good one. To see an example for a “true” SNP (or sequencing error of the GenBank reference, who knows), look at the SROc tag around position 548020.